Na temat witaminy C krąży bardzo wiele mitów i przekłamań. A ponieważ mają one w dużym stopniu oparcie w chemii, warto jest je tutaj szczegółowo rozjaśnić. Czy zatem może być tak, że witamina może być prawa i lewa? Może i to w dodatku na bardzo różne sposoby.
![]()
Kwas askorbinowy to związek szczególny. Jest kofaktorem regulującym działanie wielu enzymów. Pomaga przy produkcji kolagenu, wpływając na stan naczyń krwionośnych i skóry. Jest ważnym przeciwutleniaczem neutralizującym wolne rodniki. A przy tym jest substancją jakiej nasz organizm nie może sam wytwarzać, co zresztą stanowi wśród zwierząt wyjątek. Takie na przykład szczury same go sobie wytwarzają i nigdy nie doznają niedoboru.
Jego brak wywołuje przykre i na dłuższą metę śmiertelne choroby, jak choćby szkorbut, nazywany też obrazowo gnilcem, co zanim poznano jego rolę w żywieniu stanowiło częstą przyczyną zgonów marynarzy, pozbawionych dostępu do świeżej żywności. Nic więc dziwnego, że witamina C została nazwana kwasem a-skorbinowym, to jest antyszkorbutowym.
Człowiek wynalazł kilka prostych sposobów jego syntezy tak, aby otrzymać cząsteczkę o budowie takiej samej jak naturalna. Zwykle surowcem jest cukier glukoza, która zostaje zredukowana wodorem, poddana fermentacji przez bakterie octowe, selektywnie utleniona i odwodniona. W ten sposób, bądź metodami z większym udziałem bakterii, produkuje się ją w ilościach niemal przemysłowych i zużywa głownie jako przeciwutleniacz w żywności, środek zapobiegający brązowieniu mrożonek czy ulepszacz do pieczywa.
Zasadniczo dobrze zbilansowana dieta powinna dostarczać go nam wręcz w nadmiarze, jednak niektórzy wolą go sobie dodatkowo uzupełniać w większych dawkach. I często wpadają w pułapkę marketingu. Specjaliści od suplementów mówią im "Nie kupujcie pigułek w aptece bo w ogóle nie działają, bierzcie wyciąg z X albo tabletki dla których specjalnie potwierdzono że to jest ten właściwy, przypadkiem mamy je w ofercie. Bierz tylko nasze".
Czemu tabletkowa witamina ma nie działać? Bo "prawdziwa" i naturalna jest ta lewoskrętna, więc jeśli na opakowaniu nie zostanie wprost to napisane, to niechybnie tabletki zawierają tą nieczynną prawoskrętną.
Każdy kto ma trochę większe pojęcie o chemii, wie jak zbudowana jest cząsteczka witaminy C i wie o co chodzi z tą skrętnością, czy lewością, uśmieje się słysząc takie rzeczy. Ale niestety przeciętny konsument nie wie. Więc ja mu rzecz krótko wyjaśnię
![]()
Asymetria
Asymetria to własność obiektu, która powoduje, że obracając go w przestrzeni, odbijając w wyimaginowanym "lustrze kształtów" czy przekształcając przez punkt nie otrzymamy identycznie wyglądającej bryły. No chyba, że obrócimy go o 360 stopni czy powtórzymy odbicia dwa razy, ale to tak jakbyśmy go z miejsca nie ruszali.
Chemicy już dawno odkryli, że cząsteczki związków chemicznych, będące ułożonymi w przestrzeniami skupiskami atomów, mogą bądź posiadać jakąś symetrię, bądź nie posiadać żadnej, i to właśnie te ostatnie okazały się najciekawsze. Jeśli cząsteczka związku jest asymetryczna, to bardzo często możliwe jest, że mogą istnieć jej dwie formy, podobne do siebie jak lustrzane odbicia ale nie nakładające się na siebie.
Obiektami o takich własnościach z jakimi często mamy do czynienia, są nasze ręce - jedna dłoń jest lustrzanie podobna do drugiej, ale jedna nie nałoży się na kształt drugiej, bo kciuki odchylają się w różne strony. Jeśli złożymy dłonie jak do modlitwy sytuacja będzie podobna, bo w różne strony będą zwrócone ich grzbiety. Z tego powodu matematycy bryły o takich właściwościach, a więc posiadające lustrzanie podobne formy "lewą" i "prawą" jak dłonie, nazwali chiralnymi, od greckiego "chira" to jest ręka. (a wróżenie z dłoni to chiromancja).
Jeśli cząsteczka związku chemicznego nie będzie posiadała elementów symetrii, to także i dla niej możliwe będzie dla niej istnienie w dwóch formach, nazywanych izomerami geometrycznymi. Zazwyczaj dotyczy to związków organicznych, w których atom węgla tworzy cztery wiązania ułożone nie płasko, i wystarczy aby w którymkolwiek z węgli cząsteczki zdarzyło się, że do każdego wiązania będzie przyczepione coś innego.
![]()
Aby ten fakt opisać i jakoś odróżniać poza tym we wszystkim identyczne izomery, chemicy stworzyli szereg systemów klasyfikujących
R czy S?
Ten sposób klasyfikacji opiera się na rzeczywistej budowie związku. Aby sprawdzić jaka jest jego konfiguracja, sprowadzamy go do tego właśnie interesującego nas węgla, mającego cztery różne podstawniki, nazywanego asymetrycznym. Podstawnikom tym nadajemy pewne rangi, zależne od stopnia rozbudowania, całkowitej masy atomowej czy obecności cięższych atomów. No i otóż, jeśli ustawimy naszą cząsteczkę tak, że podstawnik najniższej rangi znajdzie się z tyłu, a trzy pozostałe będą skierowane w naszą stronę, to gdy przechodząc od podstawnika mniej ważnego do ważniejszego wykonujemy obrót w prawo, konfiguracja dla tego węgla wynosi R a gdy w lewo wynosi S.
![]() Ten sposób klasyfikacji jest bardzo ścisły, po samej nazwie możemy ustalić jak przestrzennie są poustawiane grupy wokół tego atomu.
Ten sposób klasyfikacji jest bardzo ścisły, po samej nazwie możemy ustalić jak przestrzennie są poustawiane grupy wokół tego atomu.
W jednej cząsteczce może być zawartych wiele takich atomów, ponieważ zaś każdy ma dwie możliwe konfiguracje, możliwych staje się wiele izomerów. Przykładowo glukoza ma cztery takie atomy o konfiguracji 2R,3S,4R,5R, będąc jednym z 16 możliwych izomerów aldoheksozy.
D czy L?
Klasyfikacja D/L jest używana właściwie tylko do cukrów, polialkoholi i aminokwasów, także dla witaminy C. Jest to klasyfikacja względna, w której przypisanie związku do danej kategorii odbywa się poprzez porównanie konfiguracji z pewnym wzorcem.
Tą cząsteczką wzorcową był naturalnie występujący aldehyd glicerynowy, uznany z najprostszy przypadek, składał się bowiem z trzech węgli z czym jeden tylko był asymetryczny. Klasyfikacja odbywa się następująco - ustawiamy naszą cząsteczkę aby łańcuch węglowy był ustawiony pionowo, grupa aldehydowa lub ketonowa znalazła się na górze, a grupy boczne sterczały na boki, będąc zwrócone lekko w naszą stronę (co rysuje się w ten sposób, że ich wiązania wyglądają jak czarne trójkąty):
![]() Jeśli w takim ustawieniu grupa -OH znajdzie się po prawej, to cząsteczkę zaliczymy do szeregu D a jeśli po lewej to do szeregu L. Jeśli nasza cząsteczka zawiera więcej węgli asymetrycznych, wtedy bierzemy pod uwagę tylko ten ostatni na dole.
Jeśli w takim ustawieniu grupa -OH znajdzie się po prawej, to cząsteczkę zaliczymy do szeregu D a jeśli po lewej to do szeregu L. Jeśli nasza cząsteczka zawiera więcej węgli asymetrycznych, wtedy bierzemy pod uwagę tylko ten ostatni na dole.
Jak widzicie jest to klasyfikacja bardzo arbitralna.
+ czy -?
Ostatni sposób klasyfikacji nie jest wprost związany z budową cząsteczki, a bardziej z tym jak oddziałuje ze światłem. Otóż izomery geometryczne związków wpływają na światło spolaryzowane. Jeśli przez fiolkę ze związkiem przepuścimy światło spolaryzowane przy pomocy polaryzatora ustawionego w określonym kierunku, to po przejściu przez związek kierunek polaryzacji światła trochę się przekręci. Poznajemy to po tym, że ustawiając za fiolką drugi polaryzator widzimy że część światła z fiolki jest zatrzymywana i aby uzyskać pełną przepuszczalność, musimy drugi polaryzator trochę obrócić.
Obejrzyjcie świetną demonstrację tego zjawiska dla dwóch izomerów karwonu:
I teraz najważniejsze. Uznano, że jeśli płaszczyzna polaryzacji obróciła się w prawo, to mówimy o związku że jest prawoskrętny i przypisujemy mu znaczek plus (+) a jeśli w lewo to jest lewoskrętny i przypisujemy mu znaczek minus (-). Dwa izomery optyczne tej samej substancji skręcają światło spolaryzowane o taki sam kąt w przeciwne strony, dlatego zawsze jeden jest (+) a drugi (-). Ich mieszanina pół na pół jest nieaktywna bo przeciwne oddziaływania się znoszą.
Przy czym nie koniecznie znak skręcalności powiązany jest z konfiguracją R/S czy D/L. Wprawdzie dany określony izomer geometryczny ma zawsze dany określony znak skręcalności, ale różne związki o konfiguracji D czy R mogą skręcać światło spolaryzowane w różne strony. Na przykład D-glukoza jest prawoskrętna a D-fruktoza lewoskrętna.
A jaka jest witamina?
Kwas askorbinowy zawiera dwa węgle asymetryczne, w związku z czym możliwe są dla niego cztery izomery; kwas (R,S)-L-askorbinowy, (S,R)-D-askorbinowy; (S,S)-L-izoaskorbinowy i (R,R)-D-izoaskorbinowy
![]() Aktywność biologiczną witaminy ma tylko jeden z nich, występujący naturalnie kwas (R,S) L-askorbinowy. Pozostałe nie mogą być nazywane witaminami, choć są podobnymi do niej przeciwutleniaczami..
Aktywność biologiczną witaminy ma tylko jeden z nich, występujący naturalnie kwas (R,S) L-askorbinowy. Pozostałe nie mogą być nazywane witaminami, choć są podobnymi do niej przeciwutleniaczami..
A jak wygląda czynność optyczna? Otóż będący witaminą C naturalny kwas L-askorbinowy jest prawoskrętny. Czyli ma znaczek (+).
Zatem specjaliści od wciskania ludziom suplementów, którzy twierdzą, że witamina C powinna być lewoskrętna, pewnie pomylili się widząc znaczek L mówiący o względnej konfiguracji związku. Który to mówi nam jedynie, że jeśli zapiszemy cząsteczkę związku w określony sposób to grupa -OH na ostatnim węglu asymetrycznym będzie po lewej stronie rysunku, a to nie ma nic do skrętności.
...bo jak nie napisali L to na pewno jest D
Gdy świat alternatywnej medycyny pojął wreszcie istnienie dwóch odmian kwasu askorbinowego, zaczął przekonywać, że z pewnością absolutnie ta tabletkowa witamina, to jest właśnie ta nienaturalna D. Ze jeśli nie napiszą "kwas L-askorbinowy" to znaczy, że to musi być ten drugi. Ci którzy się na to nabierają zamawiają w hurtowniach wielkie wory kwasu L, nie ufając tabletkom, no bo przecież kto wie co sobie producenci napisali.
Dlaczego to bzdura?
Jak to już powyżej napisałem, kwas L-askorbinowy produkuje się z naturalnej glukozy otrzymując właściwą "naturalną" konfigurację. Wobec tego związek jest dosyć tani i dlatego bardziej opłaca się pchać go do tabletek, niż drugi izomer, produkowany w mniejszych ilościach przy pomocy innych, bardziej skomplikowanych metod. Kwas D-askorbinowy nie występuje w naturze a organizmy nie są przystosowane do jego wytwarzania, odpada więc bardzo ułatwiający produkcję mikrobiologiczny etap syntezy. Aby go otrzymać należałoby więc albo użyć drogich katalizatorów albo szeregu przemian dających mieszaninę izomerów, którą następnie należałoby rozdzielać. Ponieważ jednak izomer D nie jest wykorzystywany w medycynie, nikt go specjalnie nie produkuje. Czemu więc tabletki miałyby zawierać nie produkowany, znacznie droższy, nieaktywny izomer, gdy w zasięgu jest produkowany na dużą skalę aktywny L-izomer?
Inna kwestia to dozwolone nazewnictwo obu odmian. Nazwa handlowa "witamina C" oraz nazwa "kwas askorbinowy" są na mocy międzynarodowych przepisów zastrzeżone tylko dla kwasu L-(+)-askorbinowego, takiego jak naturalny. Producent nie może wsadzać w preparat inny stereoizomer i nazwać go witaminą. Ponieważ w świetle przepisów jest to w sumie jasne, producent tabletek nie musi pisać na opakowaniu, że tabletka zawiera kwas L-(+)-(4R,5S)-askorbinowy abyśmy byli pewni, że jest to taka cząsteczka jak naturalna, działająca biologicznie.

Kwas askorbinowy to związek szczególny. Jest kofaktorem regulującym działanie wielu enzymów. Pomaga przy produkcji kolagenu, wpływając na stan naczyń krwionośnych i skóry. Jest ważnym przeciwutleniaczem neutralizującym wolne rodniki. A przy tym jest substancją jakiej nasz organizm nie może sam wytwarzać, co zresztą stanowi wśród zwierząt wyjątek. Takie na przykład szczury same go sobie wytwarzają i nigdy nie doznają niedoboru.
Jego brak wywołuje przykre i na dłuższą metę śmiertelne choroby, jak choćby szkorbut, nazywany też obrazowo gnilcem, co zanim poznano jego rolę w żywieniu stanowiło częstą przyczyną zgonów marynarzy, pozbawionych dostępu do świeżej żywności. Nic więc dziwnego, że witamina C została nazwana kwasem a-skorbinowym, to jest antyszkorbutowym.
Człowiek wynalazł kilka prostych sposobów jego syntezy tak, aby otrzymać cząsteczkę o budowie takiej samej jak naturalna. Zwykle surowcem jest cukier glukoza, która zostaje zredukowana wodorem, poddana fermentacji przez bakterie octowe, selektywnie utleniona i odwodniona. W ten sposób, bądź metodami z większym udziałem bakterii, produkuje się ją w ilościach niemal przemysłowych i zużywa głownie jako przeciwutleniacz w żywności, środek zapobiegający brązowieniu mrożonek czy ulepszacz do pieczywa.
Zasadniczo dobrze zbilansowana dieta powinna dostarczać go nam wręcz w nadmiarze, jednak niektórzy wolą go sobie dodatkowo uzupełniać w większych dawkach. I często wpadają w pułapkę marketingu. Specjaliści od suplementów mówią im "Nie kupujcie pigułek w aptece bo w ogóle nie działają, bierzcie wyciąg z X albo tabletki dla których specjalnie potwierdzono że to jest ten właściwy, przypadkiem mamy je w ofercie. Bierz tylko nasze".
Czemu tabletkowa witamina ma nie działać? Bo "prawdziwa" i naturalna jest ta lewoskrętna, więc jeśli na opakowaniu nie zostanie wprost to napisane, to niechybnie tabletki zawierają tą nieczynną prawoskrętną.
Każdy kto ma trochę większe pojęcie o chemii, wie jak zbudowana jest cząsteczka witaminy C i wie o co chodzi z tą skrętnością, czy lewością, uśmieje się słysząc takie rzeczy. Ale niestety przeciętny konsument nie wie. Więc ja mu rzecz krótko wyjaśnię

Asymetria
Asymetria to własność obiektu, która powoduje, że obracając go w przestrzeni, odbijając w wyimaginowanym "lustrze kształtów" czy przekształcając przez punkt nie otrzymamy identycznie wyglądającej bryły. No chyba, że obrócimy go o 360 stopni czy powtórzymy odbicia dwa razy, ale to tak jakbyśmy go z miejsca nie ruszali.
Chemicy już dawno odkryli, że cząsteczki związków chemicznych, będące ułożonymi w przestrzeniami skupiskami atomów, mogą bądź posiadać jakąś symetrię, bądź nie posiadać żadnej, i to właśnie te ostatnie okazały się najciekawsze. Jeśli cząsteczka związku jest asymetryczna, to bardzo często możliwe jest, że mogą istnieć jej dwie formy, podobne do siebie jak lustrzane odbicia ale nie nakładające się na siebie.
Obiektami o takich własnościach z jakimi często mamy do czynienia, są nasze ręce - jedna dłoń jest lustrzanie podobna do drugiej, ale jedna nie nałoży się na kształt drugiej, bo kciuki odchylają się w różne strony. Jeśli złożymy dłonie jak do modlitwy sytuacja będzie podobna, bo w różne strony będą zwrócone ich grzbiety. Z tego powodu matematycy bryły o takich właściwościach, a więc posiadające lustrzanie podobne formy "lewą" i "prawą" jak dłonie, nazwali chiralnymi, od greckiego "chira" to jest ręka. (a wróżenie z dłoni to chiromancja).
Jeśli cząsteczka związku chemicznego nie będzie posiadała elementów symetrii, to także i dla niej możliwe będzie dla niej istnienie w dwóch formach, nazywanych izomerami geometrycznymi. Zazwyczaj dotyczy to związków organicznych, w których atom węgla tworzy cztery wiązania ułożone nie płasko, i wystarczy aby w którymkolwiek z węgli cząsteczki zdarzyło się, że do każdego wiązania będzie przyczepione coś innego.

Aby ten fakt opisać i jakoś odróżniać poza tym we wszystkim identyczne izomery, chemicy stworzyli szereg systemów klasyfikujących
R czy S?
Ten sposób klasyfikacji opiera się na rzeczywistej budowie związku. Aby sprawdzić jaka jest jego konfiguracja, sprowadzamy go do tego właśnie interesującego nas węgla, mającego cztery różne podstawniki, nazywanego asymetrycznym. Podstawnikom tym nadajemy pewne rangi, zależne od stopnia rozbudowania, całkowitej masy atomowej czy obecności cięższych atomów. No i otóż, jeśli ustawimy naszą cząsteczkę tak, że podstawnik najniższej rangi znajdzie się z tyłu, a trzy pozostałe będą skierowane w naszą stronę, to gdy przechodząc od podstawnika mniej ważnego do ważniejszego wykonujemy obrót w prawo, konfiguracja dla tego węgla wynosi R a gdy w lewo wynosi S.

W jednej cząsteczce może być zawartych wiele takich atomów, ponieważ zaś każdy ma dwie możliwe konfiguracje, możliwych staje się wiele izomerów. Przykładowo glukoza ma cztery takie atomy o konfiguracji 2R,3S,4R,5R, będąc jednym z 16 możliwych izomerów aldoheksozy.
D czy L?
Klasyfikacja D/L jest używana właściwie tylko do cukrów, polialkoholi i aminokwasów, także dla witaminy C. Jest to klasyfikacja względna, w której przypisanie związku do danej kategorii odbywa się poprzez porównanie konfiguracji z pewnym wzorcem.
Tą cząsteczką wzorcową był naturalnie występujący aldehyd glicerynowy, uznany z najprostszy przypadek, składał się bowiem z trzech węgli z czym jeden tylko był asymetryczny. Klasyfikacja odbywa się następująco - ustawiamy naszą cząsteczkę aby łańcuch węglowy był ustawiony pionowo, grupa aldehydowa lub ketonowa znalazła się na górze, a grupy boczne sterczały na boki, będąc zwrócone lekko w naszą stronę (co rysuje się w ten sposób, że ich wiązania wyglądają jak czarne trójkąty):
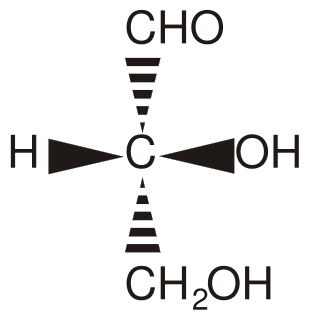
Jak widzicie jest to klasyfikacja bardzo arbitralna.
+ czy -?
Ostatni sposób klasyfikacji nie jest wprost związany z budową cząsteczki, a bardziej z tym jak oddziałuje ze światłem. Otóż izomery geometryczne związków wpływają na światło spolaryzowane. Jeśli przez fiolkę ze związkiem przepuścimy światło spolaryzowane przy pomocy polaryzatora ustawionego w określonym kierunku, to po przejściu przez związek kierunek polaryzacji światła trochę się przekręci. Poznajemy to po tym, że ustawiając za fiolką drugi polaryzator widzimy że część światła z fiolki jest zatrzymywana i aby uzyskać pełną przepuszczalność, musimy drugi polaryzator trochę obrócić.
Obejrzyjcie świetną demonstrację tego zjawiska dla dwóch izomerów karwonu:
I teraz najważniejsze. Uznano, że jeśli płaszczyzna polaryzacji obróciła się w prawo, to mówimy o związku że jest prawoskrętny i przypisujemy mu znaczek plus (+) a jeśli w lewo to jest lewoskrętny i przypisujemy mu znaczek minus (-). Dwa izomery optyczne tej samej substancji skręcają światło spolaryzowane o taki sam kąt w przeciwne strony, dlatego zawsze jeden jest (+) a drugi (-). Ich mieszanina pół na pół jest nieaktywna bo przeciwne oddziaływania się znoszą.
Przy czym nie koniecznie znak skręcalności powiązany jest z konfiguracją R/S czy D/L. Wprawdzie dany określony izomer geometryczny ma zawsze dany określony znak skręcalności, ale różne związki o konfiguracji D czy R mogą skręcać światło spolaryzowane w różne strony. Na przykład D-glukoza jest prawoskrętna a D-fruktoza lewoskrętna.
A jaka jest witamina?
Kwas askorbinowy zawiera dwa węgle asymetryczne, w związku z czym możliwe są dla niego cztery izomery; kwas (R,S)-L-askorbinowy, (S,R)-D-askorbinowy; (S,S)-L-izoaskorbinowy i (R,R)-D-izoaskorbinowy
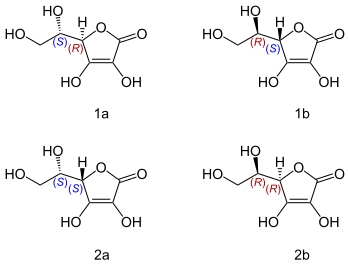
A jak wygląda czynność optyczna? Otóż będący witaminą C naturalny kwas L-askorbinowy jest prawoskrętny. Czyli ma znaczek (+).
Zatem specjaliści od wciskania ludziom suplementów, którzy twierdzą, że witamina C powinna być lewoskrętna, pewnie pomylili się widząc znaczek L mówiący o względnej konfiguracji związku. Który to mówi nam jedynie, że jeśli zapiszemy cząsteczkę związku w określony sposób to grupa -OH na ostatnim węglu asymetrycznym będzie po lewej stronie rysunku, a to nie ma nic do skrętności.
...bo jak nie napisali L to na pewno jest D
Gdy świat alternatywnej medycyny pojął wreszcie istnienie dwóch odmian kwasu askorbinowego, zaczął przekonywać, że z pewnością absolutnie ta tabletkowa witamina, to jest właśnie ta nienaturalna D. Ze jeśli nie napiszą "kwas L-askorbinowy" to znaczy, że to musi być ten drugi. Ci którzy się na to nabierają zamawiają w hurtowniach wielkie wory kwasu L, nie ufając tabletkom, no bo przecież kto wie co sobie producenci napisali.
Dlaczego to bzdura?
Jak to już powyżej napisałem, kwas L-askorbinowy produkuje się z naturalnej glukozy otrzymując właściwą "naturalną" konfigurację. Wobec tego związek jest dosyć tani i dlatego bardziej opłaca się pchać go do tabletek, niż drugi izomer, produkowany w mniejszych ilościach przy pomocy innych, bardziej skomplikowanych metod. Kwas D-askorbinowy nie występuje w naturze a organizmy nie są przystosowane do jego wytwarzania, odpada więc bardzo ułatwiający produkcję mikrobiologiczny etap syntezy. Aby go otrzymać należałoby więc albo użyć drogich katalizatorów albo szeregu przemian dających mieszaninę izomerów, którą następnie należałoby rozdzielać. Ponieważ jednak izomer D nie jest wykorzystywany w medycynie, nikt go specjalnie nie produkuje. Czemu więc tabletki miałyby zawierać nie produkowany, znacznie droższy, nieaktywny izomer, gdy w zasięgu jest produkowany na dużą skalę aktywny L-izomer?
Inna kwestia to dozwolone nazewnictwo obu odmian. Nazwa handlowa "witamina C" oraz nazwa "kwas askorbinowy" są na mocy międzynarodowych przepisów zastrzeżone tylko dla kwasu L-(+)-askorbinowego, takiego jak naturalny. Producent nie może wsadzać w preparat inny stereoizomer i nazwać go witaminą. Ponieważ w świetle przepisów jest to w sumie jasne, producent tabletek nie musi pisać na opakowaniu, że tabletka zawiera kwas L-(+)-(4R,5S)-askorbinowy abyśmy byli pewni, że jest to taka cząsteczka jak naturalna, działająca biologicznie.
































































































.jpg)